|Articles|October 5, 2018
ODC1 Gene Mutation Leads to Alopecia and Developmental Delays
Author(s)David Bai, PharmD
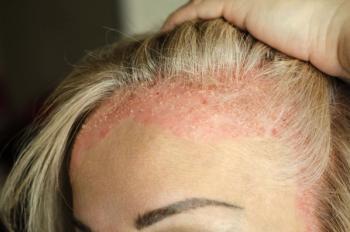
The ornithine decarboxylase 1 (ODC1) gene, for the first time in humans, has been linked to a new pediatric disorder leading to macrosomia, macrocephaly, developmental delays, alopecia, hypotonia, and hearing loss, according to a new report.
Advertisement
The ornithine decarboxylase 1 (ODC1) gene, for the first time in humans, has been linked to a new pediatric disorder leading to macrosomia, macrocephaly, developmental delays, alopecia, hypotonia, and hearing loss, according to a
ODC1 mutations associated with overexpressing c-terminally deleted ODC was described over 20 years ago in mouse models as a cause of higher polyamine (PA) biosynthesis, which plays an important role in cell developmental processes such as embryogenesis, organogenesis, and neoplastic cell growth. At the Helene DeVos Children’s Hospital, a 32-month-old female patient had sanger sequencing testing done, confirming the ODC1 variant mutation. The ODC1 mutation identified led to the deletion of the c-terminal 14 amino acid of the ODC protein and subsequently an increase in ODC protein accumulation.
At 11 months, developmental delays began to become apparent, starting with poor neck control. At 30 months, the patient remained nonverbal, making only cooing noises and occasional sound imitations. The patient was unable to sit by herself and did not have pincer grasp when reaching for objects. Spasticity in the lower extremities, nighttime eye deviation and tonic limb deviations were also observed. A direct assessment of her eyes found esotropia, bilateral myopic astigmatism, and pseudostrabismus. Nails were brittle and extremely curved, and white matter volume loss was evident on a magnetic resonance imaging (MRI) in both cerebral hemispheres. Wound healing was normal, but the patient had an unusually high tolerance for pain.
Previous studies had already identified that the c-terminal 37 amino acid of ODC led to higher ODC enzyme activity. In mice with this mutation, ODC protein and PA levels were both increased, leading to multiple hair and skin abnormalities including alopecia. In this patient, similar increases in ODC protein and PA levels were also seen. The higher PA level was also associated with increased putrescine, an amine produced when ornithine is enzymatically converted by ODC. Eflornithine (DFMO), a drug approved for use in the treatment of trypanosomiasis (African sleeping sickness), leads to putrescine production inhibition, preventing hair loss, and restoring hair growth in mice.
The use of DFMO may be justified “since some developmental delays are not likely reversible after the child has reached a certain age” said André Bachmann, a professor of pediatrics in the MSU College of Human Medicine in a news release from Michigan State University.
Bupp et al has identified the first human case of c-terminal ODC1deletion that has presented itself as a developmental disorder. DFMO is theorized as a possible therapeutic agent that can prevent neurologic deterioration and developmental delays early in the disease. Screenings for ODC1 mutations when these symptoms are present may be useful to catch and intervene other cases discovered in the future.
Reference
Bupp CP, Schultz CR, Uhi KL, Rajasekaran S, Bachmann AS. Novel de novo pathogenic variant in the ODC1 gene in a girl with developmental delay, alopecia, and dysmorphic features [published online September 21, 2018]. Am J Med Genet A.doi: 10.1002/ajmg.a.40523.
The observed patient was born at 37 weeks by cesarean section and was sent to neonatal intensive care for 35 days due to hypotonia, hypoglycemia, feeding difficulties, and hyperbilirubinemia. The patient failed the newborn hearing screen and was diagnosed with right-sided sensorineural hearing loss. Hair loss was evident almost immediately; the hair that she was born with fell out during the first month of life, and she had no eyebrows and few eyelashes.
Advertisement
Related Content
Advertisement



Advertisement
Advertisement
Trending on AJMC
1
Jeff Hunnicutt to Bring Community Oncology Roots to New Role as Navista COO
2
New MINT Trial Analysis Favors Liberal Transfusion After MI
3
Contributor: State Leaders Need Real-World Evidence to Integrate Social Care at Scale
4
Nipocalimab Improves gMG Outcomes Across East Asian Subgroup
5