




Drawing on over a decade’s worth of data, new research shows that the 10-year survival of over 9000 patients with idiopathic pulmonary arterial hypertension (IPAH) was 57%.
Drawing on over a decade’s worth of data, new research shows that the 10-year survival of over 9000 patients with idiopathic pulmonary arterial hypertension (IPAH) was 57%.

Based on their findings, the investigators argue a need for more inclusive representation of patients with pulmonary arterial hypertension (PAH) across the weight spectrum.
Repolarization dispersion has been linked to echocardiographic measures of diastolic dysfunction in past research, and a recent study found it potentially indicative of pulmonary arterial hypertension severity.

A recent study found that a selection of non-invasive markers could help identify high-risk children with pulmonary arterial hypertension associated with congenital heart disease.
A small study in northwestern China suggests earlier detection of depression and anxiety in patients with pulmonary arterial hypertension may improve patient quality of life.

Studies focused on Asian populations with pulmonary arterial hypertension (PAH) treated with sildenafil are rare, and a new meta-analysis has found the phosphodiesterase-5 inhibitor effective in this understudied subgroup.

The circulating RNA hsa_circ_0003416 appears to be downregulated in patients with pulmonary arterial hypertension (PAH), according to new research.
This new study aimed to identify metabolic differences in endothelial cells in pulmonary arterial hypertension and chronic thromboembolic pulmonary hypertension (PH) to aid in the development of novel therapeutic approaches.

High-risk patients and patients with a history of pulmonary arterial hypertension (PAH) had significantly different cytokine profiles compared with patients with a low risk of the condition.

A new report outlines helpful strategies for pediatric patients with pulmonary arterial hypertension, although experts say more research is needed to catch up with advances in the field.
Idiopathic pulmonary arterial hypertension and pulmonary hypertension due to chronic lung disease can be difficult to differentiate, but new research suggests descriptions of lung parenchyma in routine CT scans may prove useful.
Although a number of biomarkers can be used to track features of pulmonary hypertension, no single biomarker is sufficient, according to a new review.
The authors said their findings present a better option for pediatric patients with severe disease.
A review of 3 common risk assessment tools for pulmonary arterial hypertension (PAH) found they successfully predict survival for up to 5 years after initial diagnosis.
In a small study, evidence suggested 8-Hydroxy-2'-deoxyguanosine levels negatively correlated with a test of exercise endurance.
Further investigation into the mechanisms of endothelial dysfunction and its role in the pathogenesis of pulmonary arterial hypertension (PAH) may lead to more targeted therapies against the disease.

Study results regarding the clinical spectrum and inheritance pattern of GDF2 pathogenic variants suggest incomplete penetrance and/or variability of expressivity with a semi-dominant pattern of inheritance in the context of PAH.
Recent studies show that glucagon-like peptide-1 receptor agonists have anti-inflammatory effects in human and rodent pathological models, making them a potential therapeutic strategy for treating pulmonary arterial hypertension after COVID-19 infection.

This year’s most-read articles on pulmonary arterial hypertension (PAH) covered a range of topics, including nutrition, therapy switches, and patient education to improve treatment adherence. Improving patient outcomes is something they all had in common.
Drawing a clearer picture of the relationship between pulmonary artery involvement (PAI) in Takayasu arteritis (TAK), researchers have published data on the clinical features of TAK with PAI.
Pulmonary arterial hypertension is a rare form of pulmonary hypertension that poses diagnostic challenges and often comes with a delayed diagnosis.
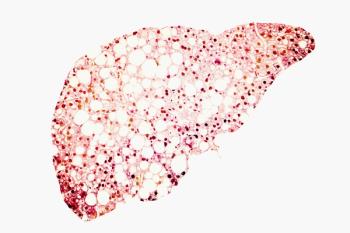
The researchers said they believe it is the first study to show the presence of hepatic fibrosis in patients with chronic right heart failure accompanied by pulmonary arterial hypertension (PAH) using liver ultrasound elastography.
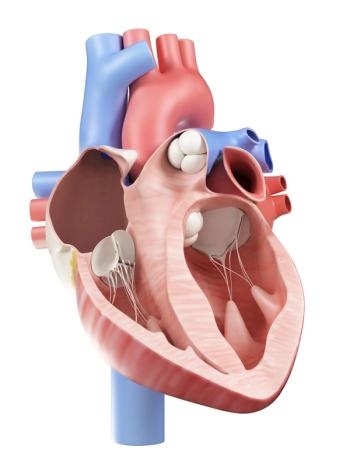
Although pulmonary arterial hypertension (PAH) is a rare occurrence in systemic lupus erythematosus (SLE), a recent report details efforts by researchers to find an easy way to identify predictive factors, since having both diseases together reduces overall survival.
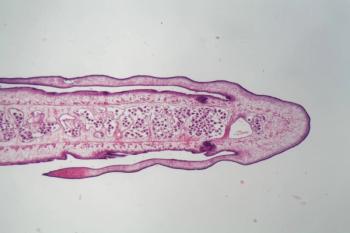
Further investigation on the pathogenesis and treatment of patients with schistosome-associated pulmonary arterial hypertension (Sch-PAH) may lead to improved symptoms and higher rates of survival.
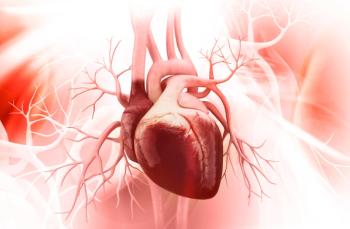
REPAIR study findings outline the benefits of macitentan for right ventricular functioning in pulmonary arterial hypertension.